Familial Hypercholesterolemia (FH) – For Clinicians
 For Clinicians (1)")
FH is the most common inherited heart disease. Heterozygous FH, which accounts for most FH cases, affects 1 in 250 people worldwide.1 Homozygous FH is rarer, affecting 1 in 160,000 to 300,000 people. Although a relatively common disease, FH is extremely under-diagnosed, with less than 10% of cases being diagnosed in many countries.2
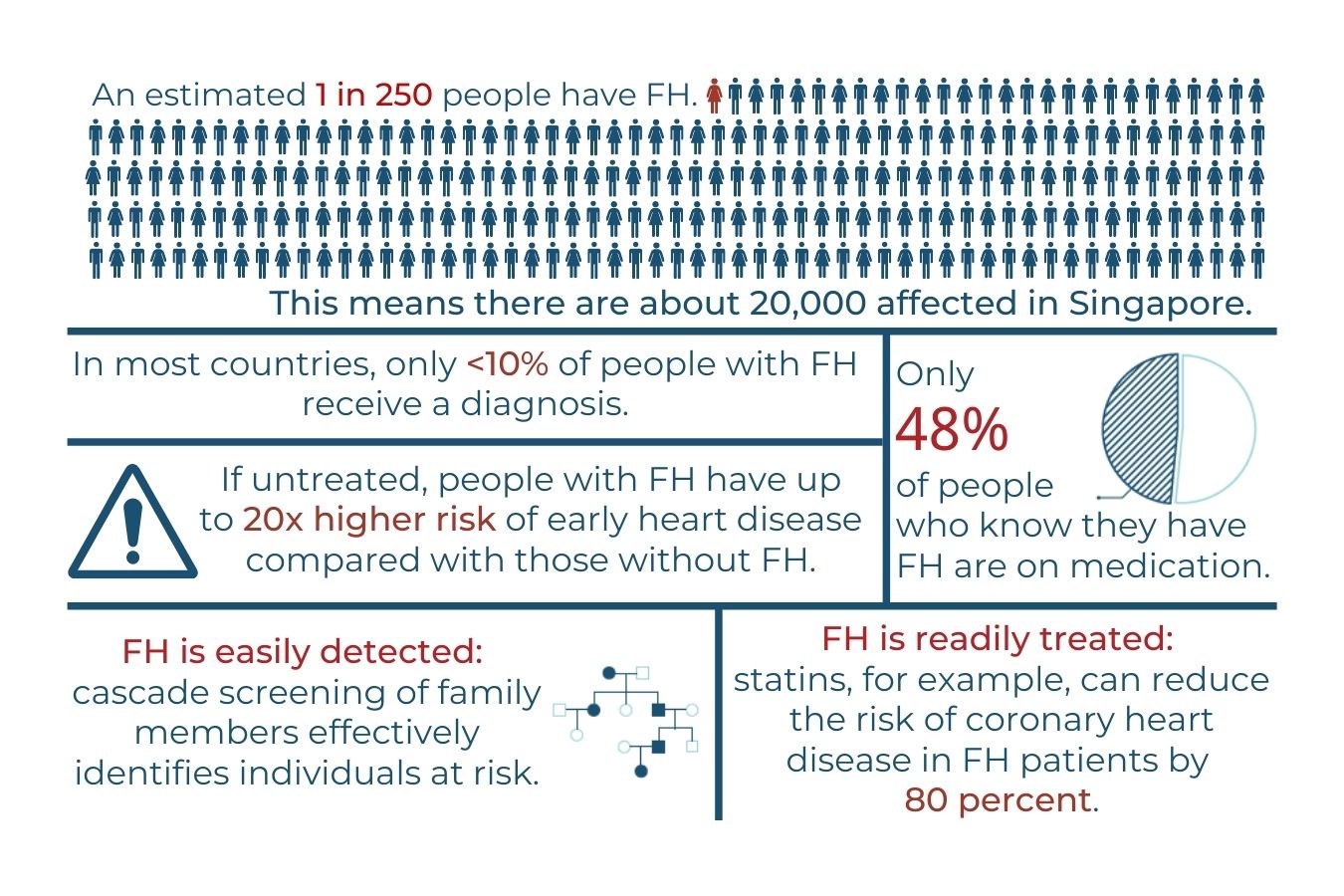
Figure 1:Key Facts and Statistics about Familial Hypercholesterolemia (FH)
What causes FH?
FH is caused by a genetic mutation, usually in one of three genes: the LDL receptor (LDLR), apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. 3 These genes code for the LDLR, APOB and PCSK9 proteins respectively (Figure 2).
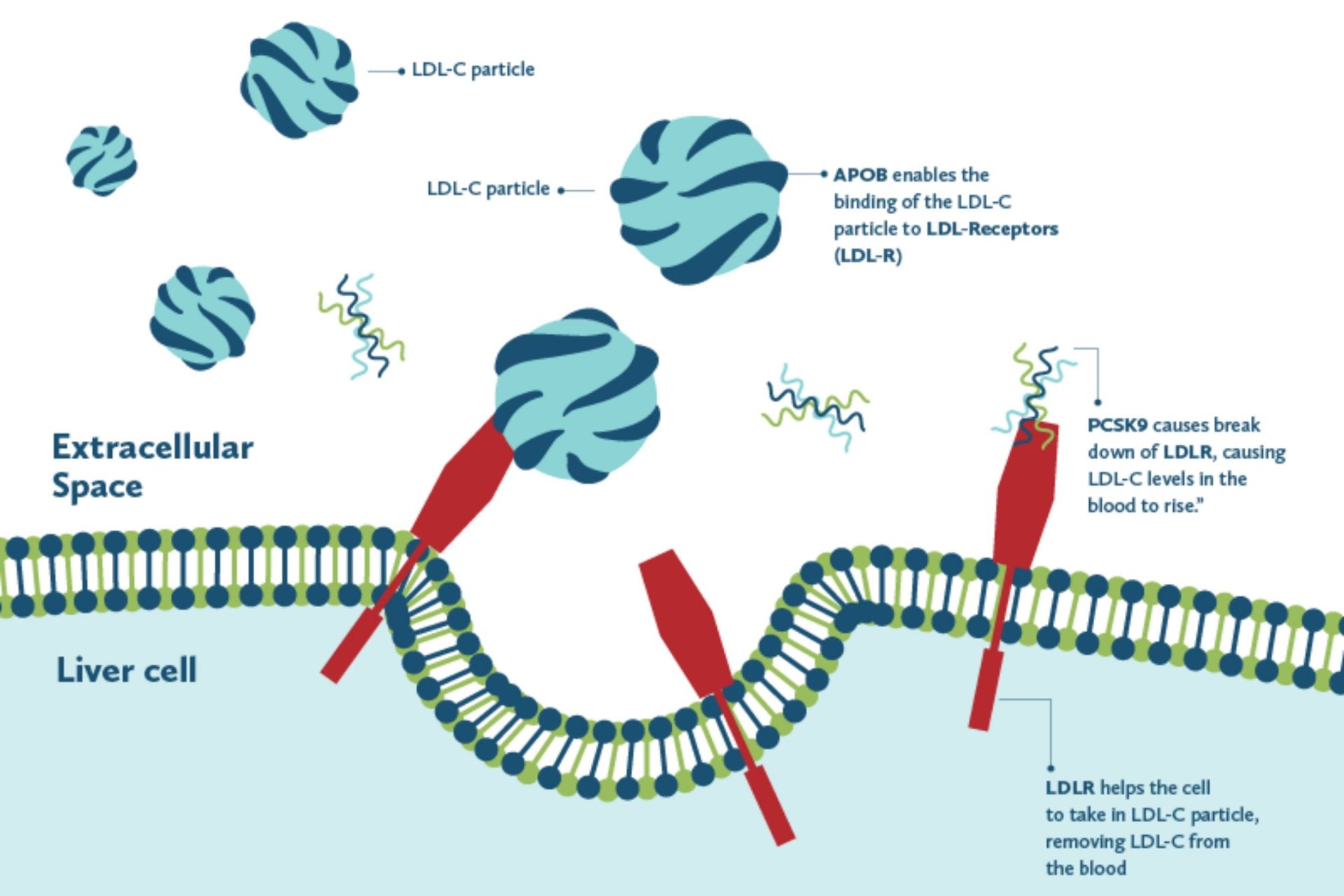
Figure 2: Effects on LDL-C particles of the proteins coded by 3 most common FH genes – APOB, PCSK9 and LDLR
Autosomal dominant inheritance
The most common FH mutations are found in the LDLR gene.
Each person inherits one LDLR gene from their mother and another LDLR gene from their father. If the child inherits one LDLR gene containing a mutation (a change that impairs the ability of the protein to take up LDL) from either parent, the child will have the most common form of FH, called heterozygous FH. In cases where both parents carry a mutation, there is a 25% chance that the child will inherit a more severe form of FH called homozygous FH. This process is called autosomal dominant inheritance.
This mechanism of inheritance, which also holds true for the APOB and PCSK9 genes, is illustrated in Figures 3A and 3B.
Most cases of FH are heterozygous, in which a patient inherits one copy of the disease-causing mutation from one of their parents.
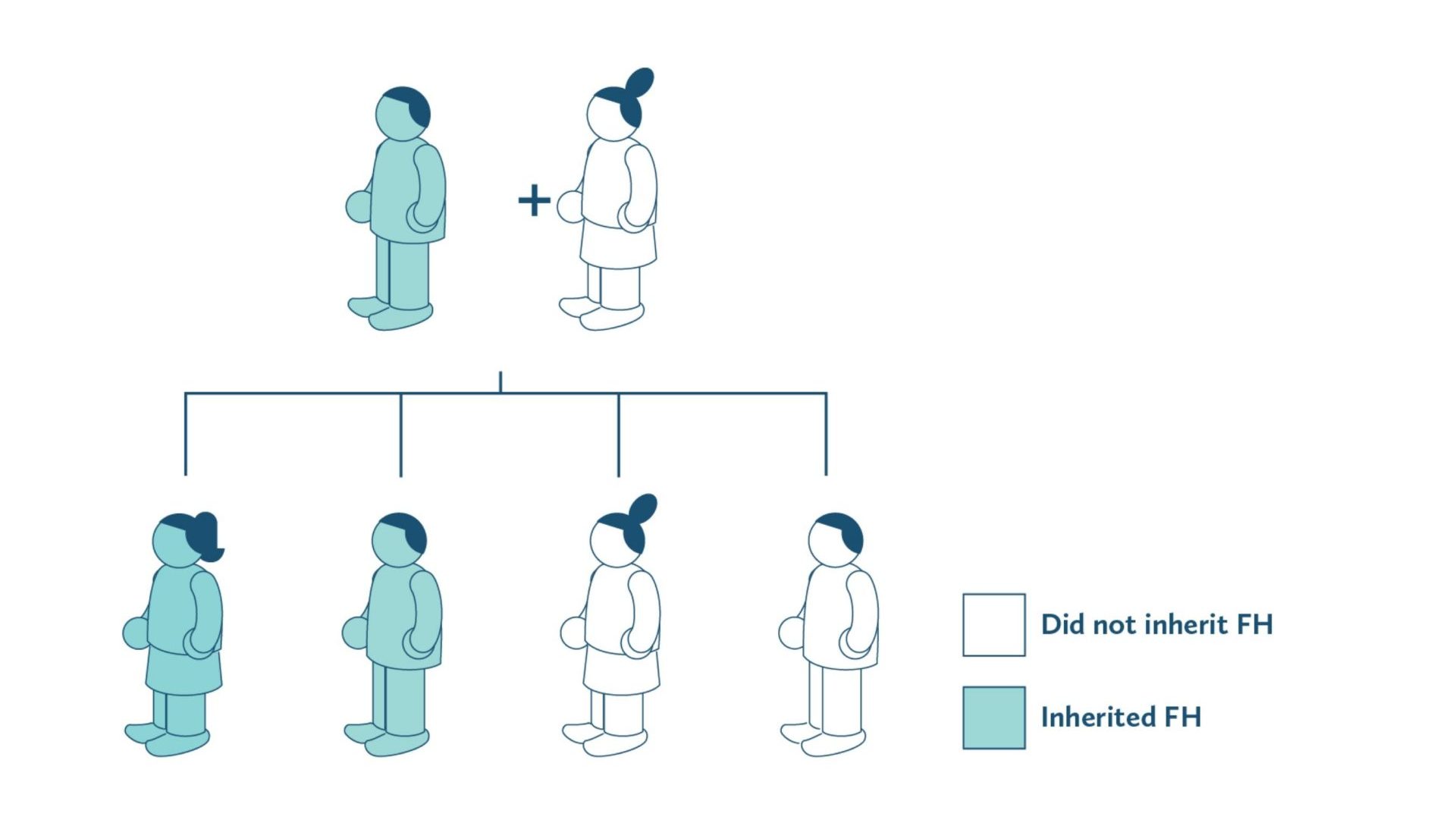
Figure 3A: Representative diagram of Heterozygous FH inheritance
A small minority of cases are homozygous, meaning they receive two copies of the mutated gene (one from each parent). In such cases, there is a 25% chance that the child will inherit a more severe form of FH.
Homozygous FH often appears in childhood and is more severe than the heterozygous form. It can even lead to heart attacks in children younger than 10. This form of FH is more difficult to treat than heterozygous FH, requiring more aggressive treatment and resulting in poorer outcomes. People with homozygous FH who are not treated rarely survive beyond 20 years of age.4

Figure 3B: Representative diagram of Homozygous FH inheritance
Autosomal recessive inheritance
A very rare form of FH, affecting 1:1,000,000 people, is passed down by autosomal recessive inheritance of a mutation in the gene coding for the LDL receptor adaptor protein 1 (LDLRAP1).5
One LDLRAP1 gene containing a mutation is inherited from each parent, impairing the function of LDLRAP1 in transporting LDL particles from the circulation into the cell. This form of FH is more severe than heterozygous FH, but generally less severe and more responsive to lipid-lowering drugs than homozygous FH. It also carries a lower risk of heart disease than homozygous FH. 5, 6
How is FH diagnosed?
- FH can be diagnosed using the Dutch Lipid Clinic Network criteria (also downloadable in pdf), which is recommended by FHCARE.7,8
[Click here to calculate LDL-C concentration for patients on treatment.] - FH can also be diagnosed using the Simon Broome Familial Hypercholesterolaemia Registry criteria9 or the Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria.10
- Alternatively, you can refer your patient to the FHCARE team, which can help establish an FH diagnosis.
Treatment for FH
Most often, statins are prescribed to lower the LDL-C concentrations in FH patients. Other medications such as ezetimibe, bile acid sequestrants and PCSK9 inhibitors may also be considered, especially for patients who cannot tolerate statins.
Along with medication, maintaining a healthy diet and weight as well as getting sufficient exercise are also important strategies for treating FH.
Despite the availability of these interventions, FH remains undertreated. Among people with an FH diagnosis, only about half are being treated with medication.
What to do if your patient has FH
If your patient has FH by the Dutch Lipid Clinic Network criteria or one of the other diagnostic criteria, you should discuss their disease risk with them and explain to them that their family members are at higher risk for FH and heart disease. Encourage your patient to ask their family members to get screened.
You may refer your patient to the FHCARE team at Khoo Teck Puat Hospital, which can confirm the diagnosis and perform genetic testing to identify the specific FH mutation. FHCARE can also perform cascade screening of your patient’s family members and provide advice on FH management.
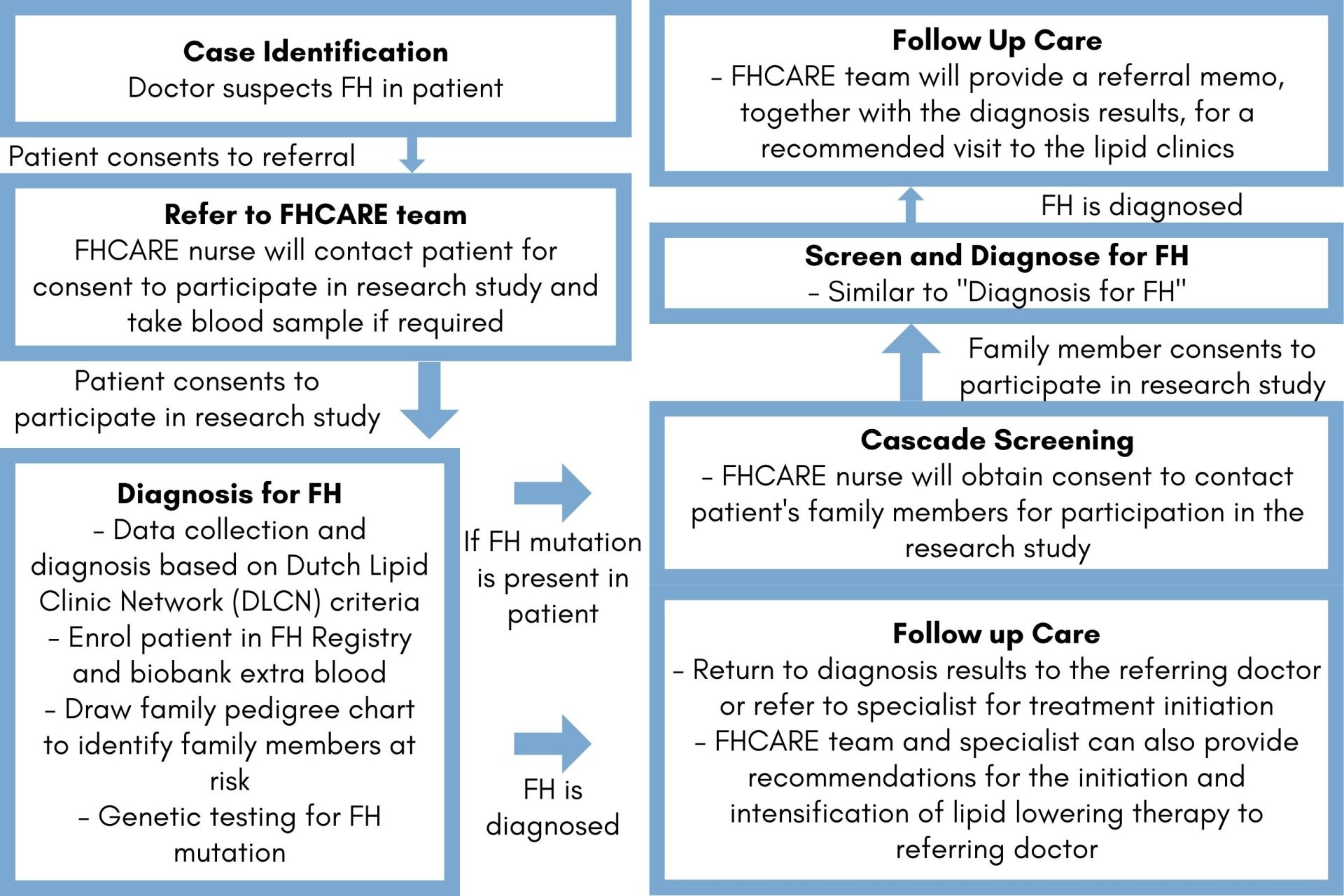
Figure 4: FHCARE workflow – Proband testing and Cascade Screening
Contact FHCARE
- E-mail: cholesterol.info@ktph.com.sg
- Phone: 6602 2346, 9674 5167, 9825 9793
Educational Materials (Downloadable)
- “Detection and Management of Familial Hypercholesterolemia” – Brochure for clinicians
- Referral Form to FHCARE (pdf)
Professional organisations and useful websites
- European Society of Cardiology: The website of this European professional organisation provides educational materials (clinical guidelines, continuing medical education [CME], webinars) on the diagnosis and treatment of cardiovascular disease (CVD), including content about FH
- American College of Cardiology: The website of this U.S. professional organisation provides educational resources, including clinical guidelines, CME materials, patient cases and clinical toolkits for CVD, including content about FH
- FH Foundation: Website of the FH Foundation, a U.S.-based non-profit organisation, housing a wide range of materials about FH for patients and clinicians
- What is FH? Heart UK: A part of the website of Heart UK, a U.K.-based non-profit organisation focused on high-cholesterol disorders
- American Heart Association: The website of this U.S. nonprofit organisation, offers many resources about heart disease and stroke, as well as materials related to FH
References
- Wiegman A, Gidding SS, Watts GF, et al; for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimising detection and treatment. Eur Heart J. 2015;36:2425-2437.
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease : Consensus Statement of the European Atherosclerosis SocietyEur Heart J. 2013;34:3478-3490.
- Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol. 2019;16:9-20.
- Sanna C, Stéphenne X, Revencu N, et al. Homozygous familial hypercholesterolemia in childhood: Genotype-phenotype description, established therapies and perspectives. Atherosclerosis. 2016 Apr;247:97-104.
- Kosmas CE, Martinez I, Morcelo R, Fabian W, Montan PD, Guzman E. Autosomal recessive hypercholesterolemia: A rare cause of familial hypercholesterolemia. Biomed J Sci & Tech Res. 2017:1.
- Pisciotta L, Oliva CP, Pes GM, et al. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): A phenotypic comparison. Atherosclerosis. 2006;188:398-405.
- World Health Organization. Familial hypercholesterolaemia. Report of a second WHO consultation. Geneva: World Health Organization; 1999.
- Civeira F; International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55-68.
- Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ. 1991;303:893-896.
- Williams RR, Hunt SC, Schumacher MC. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171-176.
 – Eng")



